Wprowadzenie
Trzcina cukrowa (Saccharum spp., Poaceae) to roślina jednoroczna lub wieloletnia uprawiana w regionach tropikalnych i subtropikalnych na całym świecie. Trzcina cukrowa jest ważną rośliną uprawną do produkcji sacharozy i stanowi 70% światowej produkcji cukru. Ponadto, jako roślina C4, trzcina cukrowa może efektywnie przekształcać energię słoneczną w energię chemiczną, a zatem jest idealną rośliną biopaliwową do produkcji etanolu (Lam i in., 2009; Santchurn i in., 2014). Rodzaj Saccharum obejmuje sześć gatunków: S. spontaneum, S. solidum, S. officinarum, S. barberi, S. sinense i S. edule, wśród których S. spontaneum jest uważany za gatunek dziki z podstawowym zestawem chromosomów x = 8. S. spontaneum jest również jednym z dwóch głównych gatunków Saccharum przyczyniających się do rozwoju współczesnych odmian trzciny cukrowej (Dhont i wsp., 1998; Ha i wsp., 1999). S. spontaneum ma silną zdolność przystosowania się do środowiska i zawiera ważne cechy genetyczne dla odporności na choroby i suszę, przyczyniając się w ten sposób do tolerancji na stres współczesnych mieszańców odmian (Grivet i in., 2004). S. spontaneum ma najszersze rozmieszczenie geograficzne, a jego poziomy ploidii wahają się od 2n = 5x = 40 do 2n = 16x = 128 (Panje i Babu, 1960).
Chromosomy trzciny cukrowej mają podobną morfologię i mały wielkość 1–6 μm na etapie skondensowanej metafazy (Ha i in., 1999; Dhont, 2005); stąd bardzo trudne jest rozróżnienie między różnymi chromosomami w oparciu o tradycyjne metody cytogenetyczne. Co więcej, jako autopoliploid, jego chromosomalne zmiany strukturalne spowodowane poliploidyzacją, duplikacją, delecją i rekombinacją są bardzo powszechne (Piperidis i in., 2010). W S. spontaneum opracowano mapy powiązań genetycznych (Silva i wsp., 1995), ale opracowanie cytologicznej mapy genetycznej pozostawało w tyle za innymi gatunkami traw. Wiarygodne narzędzia cytologiczne do identyfikacji poszczególnych chromosomów można wykorzystać do efektywnych badań genomu i wykorzystania zasobów plazmy zarodkowej, szczególnie na złożonym tle genomu współczesnych odmian trzciny cukrowej. Klony sztucznych chromosomów bakteryjnych specyficznych dla chromosomu (BAC) są nieocenionym zasobem w badaniach genomu trzciny cukrowej i będą miały wiele zastosowań w mapowaniu fizycznym, identyfikacji chromosomów i wspomaganej markerami hodowli Saccharum spp., A także w projektach sekwencjonowania i składania genomu trzciny cukrowej .
Fluorescencyjna hybrydyzacja in situ (FISH) jest potężnym narzędziem w cytologii molekularnej (Jiang i Gill, 2006). U Saccharum spp., 45S rDNA i 5S rDNA zastosowano jako sondy do wykrywania podstawowych chromosomów S. spontaneum, S. solidum i S. officinarum, w celu identyfikacji różnych podstawowych chromosomów, x = 8 w S. spontaneum i x = 10 w S. solidum i S. officinarum (Dhont i wsp., 1998; Ha i wsp., 1999). Na podstawie FISH stwierdzono, że współczesne odmiany trzciny cukrowej zawierają 70–80% chromosomów pochodzących z S. officinarum i 10–23% chromosomów z S. spontaneum, podczas gdy 5–17% wydaje się być produktem rekombinacji między S. spontaneum i S. officinarum (Dhont i wsp., 1996; Piperidis i wsp., 2001; Cuadrado i wsp., 2004). Co więcej, eliminacja chromosomów, rekombinacja i translokacja zostały wykryte u niektórych przodków pochodzących z hybrydyzacji między Saccharum i Erianthus arundinaceus przy użyciu FISH (Dhont i wsp., 1995; Huang i wsp., 2015).
Sorghum ma mały diploidalny genom (730 Mbp) z niską częstotliwością rekombinacji chromosomów. Sorgo ma wysoką syntezę z genomem trzciny cukrowej, co czyni go idealną rośliną referencyjną do analiz porównawczych z genomem trzciny cukrowej (Ming i in., 1998; Nazeema i in., 2007; Wang i in., 2010; Aitken i in., 2014 ). W tym badaniu, w oparciu o dostępne sekwencje dla zasobów BAC o wysokim pokryciu, opracowano niezawodne sondy BAC do identyfikacji chromosomów FISH i konstrukcji mapy cytogenetycznej. Celem tego badania było: (1) opracowanie zestawu sond BAC-FISH specyficznych dla chromosomu na chromosomie S. spontaneum oraz (2) zbadanie rearanżacji chromosomu między S. spontaneum i sorgo.
Materiały i metody
Materiały
Saccharum spontaneum SES208 (2n = 8x = 64) zastosowano do analiz cytologicznych w tym badaniu. Rośliny SES208 wyhodowano na polu kampusu Uniwersytetu Rolniczego i Leśnego w Fujian (Fuzhou, Chiny) w lutym 2015 r. I utrzymywano w warunkach regularnego wzrostu trzciny cukrowej.
Z haploidalnego genomu skonstruowano bibliotekę BAC S. spontaneum AP85-441 (4x = 32), który pochodzi z S. spontaneum SES208 (2n = 8x = 64) poprzez hodowlę pylników in vitro. Rozmiar genomu AP85-441 wynosi około 3,2 Gbp. Biblioteka składała się z 38 400 klonów, ze średnią wielkością insertu 100 kb, obejmującą 6x całego genomu.Ta biblioteka BAC została wykorzystana do identyfikacji transporterów sacharozy i rodzin genów fruktokinazy w S. spontaneum (Zhang i in., 2016; Chen i in., 2017).
Screening the BAC Library
35,156 klonów BAC z bibliotek AP85-441 połączono w 701 bibliotek. Każda biblioteka zawiera średnio 50 klonów BAC. Biblioteki DNA przygotowano za pomocą zestawu PhasePrepTM BAC DNA Kit (Sigma, Stany Zjednoczone) zgodnie z protokołami producenta. Biblioteki DNA BAC zsekwencjonowano przy użyciu platformy Illumina HiSeq 2500 z modelem PE250. Zsekwencjonowano łącznie 686 bibliotek (18 bibliotek, których sekwencjonowanie nie powiodło się) i wygenerowano 267,5 Gb oczyszczonych danych po przycięciu przy użyciu Trimmomatic w wersji 0.36 (Bankevich i wsp., 2012). Dane z każdej puli BAC zostały zebrane przy użyciu SPAdes w wersji 3.09 z domyślnym parametrem. W sumie 2611145 kontigów złożono z kontigiem N50 o wielkości 7,38 kbp (Zhang i Ming, dane niepublikowane).
Sekwencje biblioteki BAC AP85-441 przeszukano metodą BLAST względem genomu sorgo z wartością E 1e -4. Sekwencje, które miały pojedyncze trafienia BLAST w genomie sorgo wybrano jako kandydujące sondy FISH. Aby zidentyfikować klony BAC o niskiej liczbie kopii z biblioteki BAC, zaprojektowano startery specyficzne dla sekwencji przy użyciu oprogramowania Primer 5.0, aby umożliwić badanie przesiewowe pul BAC za pomocą PCR. Wybrano długości starterów 18–25 pz, wartości Tm 55–65 ° C i skład nukleotydów 40–60% cytozyny i guaniny. Do przeszukiwania puli wymiarów 3D biblioteki BAC zastosowano dwuetapową metodę PCR (Asakawa i wsp., 1997; Crooijmans i wsp., 2000) z pewnymi modyfikacjami. Każdy klon z 384-dołkowej płytki hodowano w 80 μl bulionu Lysogeny + 34 mg / ml chloramfenikolu przez noc w 37 ° C na 384-dołkowej płytce do hodowli. W sumie wyhodowano osiem 384-dołkowych płytek z łącznie 3072 klonami BAC. Dla każdej 384-dołkowej płytki najpierw skonstruowaliśmy 16 rzędów pul (rzędy od A do P) i 24 pule kolumnowe (kolumny 1–24) dla każdej 384-dołkowej płytki, mieszając równe objętości hodowli każdego klonu, a następnie 16 pul wierszowych i 24 pule kolumn wraz z równymi objętościami z każdego rzędu i puli kolumn jako jedna pula płytowa lub superpula. W tym badaniu skupiliśmy się na badaniu przesiewowym ośmiu 384-dołkowych płytek. W ten sposób przygotowano osiem superpuli oddzielnie dla ośmiu płyt. Zidentyfikowano pozytywną 384-studzienkową płytkę z ośmiu płytek, a następnie przeszukano dodatni rząd spośród 16 mieszanin. Ostatecznie kolumny pozytywne pod względem PCR wybrano spośród 24 mieszanin kolumn. Przeprowadzono dwadzieścia cztery reakcje PCR w celu zidentyfikowania pozytywnych klonów z ośmiu 384-dołkowych płytek.
Amplifikację PCR przeprowadzono zgodnie z wcześniejszym opisem (Bouzidi i wsp., 2006). Każda reakcja zawierała premiks rTaq 7,5 μl, 0,6 μl każdego startera (10 μM), 1,0 μl płynnej matrycy bakterii, do końcowej objętości 20 μl. Po wstępnej denaturacji w 95 ° C przez 5 min wykonano 35 cykli 95 ° C przez 30 s, 55 ° C przez 30 s i 72 ° C przez 1 min. Produkty PCR analizowano na 1,5% żelu agarozowym.
Przygotowanie chromosomu metafazowego
Przygotowanie chromosomu przeprowadzono zgodnie z wcześniejszym opisem (Lou i wsp., 2010) z niewielkimi modyfikacjami. W skrócie, rośliny S. spontaneum hodowano w warunkach szklarniowych do zbioru wierzchołków korzeni. Wycięte wierzchołki korzeni około 1–2 cm traktowano 0,002 mol / l 8-hydroksychinoliny w temperaturze pokojowej przez 2–4 h, płukano w wodzie przez 15 min i utrwalano w etanolu: kwasie octowym (3: 1) przez co najmniej 24 godz. hw temperaturze pokojowej. Końcówki korzeni następnie trawiono w roztworze enzymu (4% celulozy R-10 i 2% pektolizy w 0,1 M buforze cytrynianowym) w 37 ° C przez 1 godzinę, przemywano wodą dejonizowaną z lodem przez 30 minut i na koniec inkubowano w etanolu: kwas octowy (3: 1) przez około 30 min. Szkiełka przygotowano zgodnie z metodą „suszenia płomieniowego” (Iovene et al., 2008).
Oczyszczanie DNA BAC i znakowanie sond
DNA BAC ekstrahowano za pomocą PhasePrepTM BAC DNA Kit zgodnie z instrukcją producenta. Oczyszczone DNA BAC znakowano za pomocą standardowej reakcji translacji nici, w tym rozcieńczonej DNazy I, buforu do translacji 10 x nick, polimerazy DNA I, dNTP, dUTP znakowanych biotyną / digoksigeniną i DNA BAC. Mieszanina inkubowano w 15 ° C przez 1,5 h. Następnie pocięte produkty badano na 1,5% żelu agarozowym pod kątem obecności rozmazu między 300 a 500 pz. Otrzymane sondy przechowywano w temperaturze -20 ° C do momentu użycia.
Hybrydyzacja i wykrywanie in situ
Mieszanie i hybrydyzacja sond: Najpierw mieszanina sond (50% dejonizowanego formamidu, 2 × SSC, 80 ng DNA znakowanego digoksygeniną / biotyną, 10% siarczanu dekstranu , > 1 μg C ° t-100) umieszczono w gorącym bloku o temperaturze 90 ° C na 5 min, a następnie natychmiast przeniesiono na lód do czasu, gdy sonda była gotowa do hybrydyzacji na. Przygotowane wcześniej suszone płomieniowo szkiełka zostały poddane działaniu 70% dejonizowanego formamidu, denaturowane na bloku grzejnym w 80 ° C przez 1.5 minut, natychmiast zanurzone kolejno w lodowatym 70% etanolu, 90% etanolu, a następnie 100% etanolu na 5 minut, a następnie wysuszone na powietrzu na stole laboratoryjnym. Na koniec do każdego szkiełka dodano denaturowane sondy i przykryto szkiełkami nakrywkowymi 24 × 32 mm. Szkiełka umieszczono w wilgotnej komorze w 37 ° C na noc.
Wykrywanie sondy: szkiełka nakrywkowe usunięto i szkiełka przemyto w temperaturze pokojowej w 2 x SSC przez 5 min, w 42 ° C przez 2 x SSC przez 10 min i w temperaturze pokojowej w 1 x PBS przez 5 min w tej kolejności. Sygnały sondy znakowanej biotyną wykryto za pomocą 2 mg / ml streptawidyny Alexa Fluor 488, a sygnały sondy znakowanej digoksygeniną wykryto za pomocą 2% anty-digoksigeniny-rodaminy z owiec. Koktajl przeciwciał (100 μl buforu TNB, 1 μl Alexa Fluor 488 streptawidyny, 1 μl rodaminy anti-dig-owiec) dodano do szkiełek, które przykryto szkiełkami nakrywkowymi 24 × 32 mm, inkubowano przez 1 hw 37 ° C w wilgotną komorę i przemywano w temperaturze pokojowej w 1 x PBS trzy razy po 5 min. Nadmiar cieczy usunięto i dodano 4 ', 6-diamidyno-2-fenyloindol (DAPI) (Sigma, St. Louis, MO, Stany Zjednoczone) w roztworze przeciwdziałającym tworzeniu się farby Vectashield (Vector, Burlingame, CA, Stany Zjednoczone) w celu wybarwienia kontrastowego chromosomy. Obrazy wykonano za pomocą mikroskopu epifluorescencyjnego Olympus BX63. Obrazy sygnału FISH przeanalizowano za pomocą oprogramowania CellSens Dimension.
Wyniki
Badanie przesiewowe klonów BAC o niskiej liczbie kopii
Do przesiewania klonów BAC, które potencjalnie mają niską liczbę kopii sekwencje w genomie S. spontaneum, przeszukaliśmy za pomocą BLAST sekwencjonowane biblioteki BAC AP85-441 względem genomu sorgo. Przebadano łącznie 2000 sekwencji BAC odpowiadających 10 chromosomom sorgo o dobrej kolinearności i wykazujących swoistość sekwencji. Sekwencje BAC 2000 AP85-441 zostały zamaskowane za pomocą REPEATMASKER względem bazy danych DNA sorgo o dużej liczbie powtórzeń i bazy danych TIGR gramineae w celu przefiltrowania sekwencji powtórzeń. Ostatecznie do analizy FISH wybrano 114 BAC rozmieszczonych na 10 chromosomach sorgo (tabela uzupełniająca 1) z 7–16 BAC na każdym z 10 chromosomów sorgo (ryc. 1). W celu przeszukania pozytywnych klonów BAC zaprojektowano 114 specyficznych starterów w oparciu o sekwencje klonów BAC (tabela uzupełniająca 2), a 49 pozytywnych klonów BAC przeszukano z bibliotek BAC (tabela uzupełniająca 3).
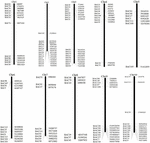
RYSUNEK 1. Rozkład 114 klonów BAC z AP85-441 w chromosomach sorgo. BAC1-114 reprezentuje liczbę BAC, dane po prawej stronie to fizyczne położenie BAC w odpowiednim regionie chromosomów sorgo. Jednostka = bp.
Siła i dystrybucja sygnału BAC-FISH
Próbki DNA wyizolowane z 49 dodatnich Klony BAC znakowano biotyną lub digoksigeniną. Sondy 5S rDNA i 45S rDNA zastosowano jako kontrolę i hybrydyzowano z chromosomami metafazy somatycznej S. spontaneum zgodnie z procedurą FISH. Ze względu na korelację między siłą sygnału a zmiennością zawartości powtarzanych sekwencji sekwencji BAC, jako konkurenta zastosowaliśmy C ° t-100 (Tabela 1). Do dalszej analizy FISH wybrano klony BAC wykazujące silne i stabilne sygnały hybrydyzacji.

TABELA 1. Klasyfikacja 27 pozytywnych klonów z blokującym DNA.
W sumie można zaobserwować 64 odrębne chromosomy w metafazie S. spontaneum. Każdy BAC-FISH przeprowadzono w czterech niezależnych eksperymentach (lub slajdach). W każdym szkiełku przeanalizowano co najmniej 10 rozprzestrzeniania się chromosomów metafazy somatycznej. Wyniki pokazały, że 27 specyficznych sond (tabela uzupełniająca 4) można podzielić na pięć grup na podstawie sygnału hybrydyzacji (tabela 1). W grupie I (w tym BAC-18, BAC-20, BAC-29 i BAC-84) sondy wykazywały osiem wyróżniających się miejsc bez konkurencyjnej supresji in situ (CISS) przy użyciu C ° t-100 (Rysunek 2A), co sugeruje sekwencje istniały w tych sekwencjach BAC. W grupie II (w tym BAC-2, BAC-11, BAC-14, BAC-19, BAC-31, BAC-33, BAC-43, BAC-71, BAC-73 i BAC-74), C ° t -100 zastosowano jako konkurenta do hybrydyzacji sondy BAC, aw kariotypach tej grupy wykazano sześć do siedmiu całkiem różnych miejsc (Figura 2B). Wynik hybrydyzacji był podobny do 45S rDNA z siedmioma miejscami sygnałowymi, co może być spowodowane zmiennością struktury chromosomu w chromosomie homologicznym. W grupie III (w tym BAC-26, BAC-66, BAC-76 i BAC-78) DNA C ° t-100 zastosowano jako konkurent i można było zaobserwować w kariotypie osiem różnych miejsc, co sugeruje tę grupę BAC klony mają powtarzalne sekwencje DNA (ryc. 2C). W grupie IV (w tym BAC-24) obserwowano więcej niż osiem miejsc sygnałowych w kariotypie i pojawiły się one w regionach pericentromerycznych i telomerycznych (ryc. 2D), co sugeruje, że sondy BAC miały powtarzalne sekwencje w S. spontaneum.W grupie V (w tym BAC-4, BAC-5, BAC-32, BAC-34, BAC-35, BAC-69, BAC-77 i BAC-81) sondy wykazywały sygnały rozproszone we wszystkich chromosomach z ograniczonymi regionami poszczególnych chromosomów wyświetlających określone sygnały (Rysunek 2E). Wynik hybrydyzacji wskazał, że ta grupa klonów BAC zawierała duże ilości powtarzalnego DNA w S. spontaneum, co może być spowodowane szybką dywergencją ewolucyjną powtarzających się regionów po podziale sorgo i S. spontaneum. Dlatego z 49 sond BAC zestaw 19 sond BAC (tabela uzupełniająca 3) z grup I, II i III wykazał specyficzne sygnały na chromosomach S. spontaneum odpowiadające dziewięciu chromosomom sorgo obok Sb05. Sygnały z pozostałych 22 sond były rozproszone we wszystkich chromosomach. Wybraliśmy zestaw 19 sond BAC jako sygnały specyficzne dla chromosomu S. spontaneum (Ryc. 3). Z 19 sond BAC jedna sonda była zlokalizowana na Sb02, Sb04 i Sb09; dwie sondy na Sb07, Sb08 i Sb10; trzy sondy na Sb01 i Sb06; i cztery sondy na Sb03. Ten zestaw sond zapewnił przydatne narzędzie do analizy porównawczej S. spontaneum i sorgo (Tabela 2).
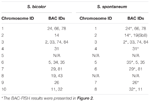
TABELA 2. Rozkład sond BAC-FISH w chromosomach S. bicolor i S. spontaneum.
Budowa biblioteki BAC specyficznej dla chromosomów S. spontaneum
Sb01 i Sb02 to dwa największe chromosomy sorgo. Aby wykryć syntezę genomu między sorgo i S. spontaneum, wybrano 10 klonów BAC specyficznych dla chromosomu z grup I, II i III. Z tych 10 sond BAC, trzy sondy (ID klonu BAC: 24, 66, 78) były homologiczne do Sb01, a siedem sond (ID klonu BAC: 14, 18, 20, 69, 71, 73, 77) odpowiadało Sb02. Wykorzystaliśmy dwukolorowe wykrywanie FISH dla komplementarnie znakowanych par BAC oraz sondy do mapowania cytogenetycznego związku łańcucha dla klonów BAC. W ten sposób stworzyliśmy mapę cytogenetyczną S. spontaneum na podstawie 10 sond BAC.
Dla trzech sond odpowiadających Sb01, analiza FISH S. spontaneum (SES208) wykazała, że BAC-24 i BAC -78 sond znajdowało się głównie w dystalnym regionie chromosomu 1 S. spontaneum, a BAC-66 mapowano w bliskim sąsiedztwie regionu centromerowego (ryc. 4). BAC-24 i BAC-66 zlokalizowane na tym samym chromosomie (chromosom 1 S. spontaneum); jednocześnie, podczas gdy sonda 66 i sonda 78 również znajdują się na tym samym chromosomie, co pokazuje, że trzy sondy BAC są rozmieszczone na tych samych chromosomach w S. spontaneum, co sugeruje syntezę między S. spontaneum i sorgo dla chromosomu 1. Wyniki FISH wykazały, że siedem sond BAC dopasowane do Sb02; tylko sondy 69, 71, 73 i 77 z siedmiu BAC generowały intensywne sygnały na jednym chromosomie S. spontaneum (SsChr2) (Figura 5); podczas gdy sondy 14, 18 i 20 były niewykrywalne na chromosomie S. spontaneum 2, ale były obserwowane na innych chromosomach S. spontaneum. Wyniki te pokazały, że rearanżacje chromosomów wystąpiły w odpowiednim Sb02 między S. spontaneum a sorgo.
Co więcej, sekwencja BAC-2 wykazuje wysokie podobieństwo (97%) do fragmentu na chromosomie 3 sorgo. Dwukolorowa analiza FISH wykazała, że zarówno BAC-2, jak i 45s rDNA zostały zmapowane do tej samej lokalizacji chromosomu S. spontaneum (Ryc. 6).
Przegrupowanie chromosomów na Sb02 między Sorgo i S. spontaneum
Aby dalej badać przegrupowanie chromosomów na Sb02 między sorgo i S. spontaneum, chromosom -specyficzne sondy odpowiadające Sb02 (BAC-14, BAC-18 i BAC-20) i odpowiadające Sb08 (BAC-19 i BAC-43) zastosowano do analizy FISH. Wyniki pokazały, że BAC-14 i BAC-19 zostały zmapowane do różnych ramion tych samych chromosomów S. spontaneum, co wskazuje, że rearanżacje chromosomów miały miejsce między chromosomami 2 i 8 w S. spontaneum i sorgo (ryc. 7).
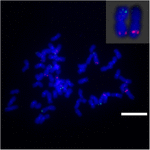
RYSUNEK 7. Przegrupowanie chromosomu trzciny cukrowej i sorgo ujawnione przez FISH. Sondy BAC-14 i BAC-19 znakowane biotyną (kolor zielony) i digoksigeniną (kolor czerwony). Słupki skali = 10 μm.
Dyskusja
Saccharum to złożony rodzaj charakteryzujący się wysokim poziomem poliploidii, z małymi chromosomami (Ha i wsp., 1999; Dhont, 2005). Rozróżnienie poszczególnych chromosomów Saccharum na podstawie ich morfologii jest bardzo trudne. Korzystając z sekwencji, które są w dużym stopniu pokryte przez bibliotekę BAC S. spontaneum, byliśmy w stanie przeszukać potencjalne klony BAC o niskiej liczbie kopii w S. spontaneum, wykorzystując genom sorgo jako odniesienie. Jako gatunek o wysokiej poliploidalności, S. spontaneum może zawierać więcej rearanżacji genomu niż dyploidy.Różnice w genomach między S. spontaneum i sorgo mogą być przyczyną niedokładności w przewidywaniach sekwencji BAC o niskiej liczbie kopii w genomie S. spontaneum. W tym badaniu początkowo uzyskaliśmy 114 potencjalnych sekwencji BAC z pojedynczą kopią opartą na genomie sorgo. Jednak tylko 49 można było zidentyfikować w częściowych bibliotekach BAC, a 27 uznano za sondy BAC-FISH specyficzne dla chromosomu. Istnieją co najmniej dwa powody, aby wyjaśnić te wyniki. Po pierwsze, 114 sekwencji BAC stanowiły częściowe fragmenty insercji BAC z powodu ograniczeń składania sekwencji NGS i mogą zawierać sekwencje powtórzone w innych regionach, które nie mają informacji o sekwencji. Te potencjalnie odkryte sekwencje powtórzeń mogą wprowadzać w błąd wykrywanie sekwencji o niskiej liczbie kopii poprzez porównanie sekwencji. Po drugie, niedawna poliploidyzacja w S. spontaneum może powodować zmienność sekwencji powtórzeń między S. spontaneum i sorgo, a tym samym skutkować niespecyficznym sygnałem FISH w S. spontaneum. W poprzednim badaniu sekwencjonowanie 20 BAC w hybrydach trzciny cukrowej wygenerowało sekwencje kontigowe o wielkości 1,45 Mb, sekwencje zrównane z genomem sorgo miały rozpiętość 0,99 Mb, co stanowiło około połowy sekwencji BAC trzciny cukrowej (Wang et al., 2010). Oczywiście delecje / insercje istniały między sorgo i Saccharum, a wykorzystanie genomu sorgo jako odniesienia nie może zastąpić unikalnych cech genomu Saccharum, a tym samym może spowodować nieprzewidziane wyniki FISH.
Niektóre slajdy FISH miały silne szum tła, który może być spowodowany małymi regionami sekwencji powtórzeń (takich jak sekwencje SSR) w klonie BAC, które w konsekwencji powodują interferencję podczas hybrydyzacji. Genom sorgo zawiera około 61% powtórzeń (Paterson i in., 2009), podczas gdy około połowa genomu składa się z sekwencji powtórzeń w hybrydach Saccarrum (De i in., 2014). W tych bogatych w powtórzenia genomach trudno jest opracować specyficzne dla sygnału sondy FISH, które cytogenetycznie rozróżniają chromosomy o podobnej morfologii. CISS z blokującym DNA C ° t-100 może skutecznie wykluczyć powtórzenie sekwencji. W tym badaniu blokujący DNA C ° t-100 wykorzystano do analizy FISH sond BAC w grupach II i III (ryc. 2D – F), które zweryfikowano jako sondy BAC specyficzne dla chromosomu, natomiast sondy FISH sond BAC w grupach IV i V powodował silne zakłócenia tła (rysunki 2D – F). Oczywiste jest, że wśród badanych sond BAC występują odmiany sekwencji powtórzeń. Dlatego też optymalizacja eksperymentów byłaby konieczna do analizy BAC-FISH w Saccharum. Sześć do siedmiu miejsc zostało wyświetlonych w grupie II, co mogło być spowodowane zmianami w strukturze homologicznych chromosomów, takimi jak delecja fragmentu w jednym lub dwóch homologicznych chromosomach. W pszenicy heksaploidalnej BAC 676D4 hybrydyzował silniej z chromosomami genomu A niż z chromosomami genomu B i D (Zhang i wsp., 2004). Te niewykrywalne od jednego do dwóch sygnałów w homologicznych chromosomach mogą być również spowodowane problemami technicznymi BAC-FISH dla tak wielu chromosomów o niewielkich rozmiarach.
W tym badaniu zidentyfikowaliśmy 27 specyficznych chromosomów BAC- Sondy FISH, które odpowiadają 9 z 10 chromosomów sorgo (wszystkie oprócz Sb05). W badaniu mapy genetycznej uzyskanej ze skrzyżowania S. officinarum z odmianą trzciny cukrowej, Sb05 połączono z Sb06 w trzcinie cukrowej (Aitken et al., 2014). W naszym badaniu mapa genetyczna oparta na populacji F1 S. spontaneum ujawniła, że Sb05 został podzielony na dwa segmenty, a dwa segmenty zostały połączone odpowiednio z Sb06 i Sb07 (Zhang i Ming, dane niepublikowane). Niedawno mapa genetyczna przedstawiciela Andropogoneae, Miscanthus sinensis, wykazała, że Sb05 ma słabą kolinearność z odpowiednią grupą sprzężeń u M. sinensis (Ma i in., 2012). Podobnie dwa pradawne chromosomy kukurydzy ortologiczne względem Sb05 zachowują najmniejszą liczbę syntenicznych ortologów genów sorgo (Schubert i Lysak, 2011). Dlatego brak BAC-FISH odpowiadającego Sb05 może wskazywać na fuzję chromosomów w S. spontaneum.
Sorgo i S. spontaneum rozeszły się 12 milionów lat temu (MYA), podstawowa liczba chromosomów została zmniejszona z x = 10 do x = 8. W Aitken i in. (2014), HG2 (grupa homologiczna 2) trzciny cukrowej dopasowana do Sb05 i Sb06 oraz HG8 (grupa homologiczna 8) do Sb02 i Sb08, dostarczając dowodów na zdarzenie podstawowej redukcji chromosomów (Aitken et al., 2014). W tym badaniu zaobserwowaliśmy, że chromosomy sorgo Sb02 i Sb08 miały międzychromosomalną rearanżację w S. spontaneum, jak wykazano na podstawie dowodów, że BAC-14 jest wyrównany z Sb02, a BAC-19 wyrównany z Sb08 (Rysunek 7). Nasze badanie dostarczyło pierwszych fizycznych i cytogenetycznych dowodów na rearanżację między chromosomami sorgo u S. spontaneum. Niepublikowane dane z naszej grupy ujawniły również, że Sb08 jest podzielony na dwa segmenty i zostały połączone odpowiednio z Sb02 i Sb09 w S. spontaneum (Zhang i Ming, dane niepublikowane).Sondy odpowiadające Sb09, Sb08, Sb05 i Sb06 można dalej stosować do badania zdarzeń między chromosomami między sorgo i S. spontaneum. S. spontaneum ma szeroki zakres poziomów ploidii (2n = 40–128) (Irvine, 1999). Te sondy BAC mogą być użyte do potwierdzenia między chromosomowych przegrupowań S. spontaneum z różnymi poziomami poliploidii.
Ze względu na różną liczbę podstawowych chromosomów między sorgo (x = 10) a S. spontaneum (x = 8) chromosomy sorgo nie odpowiadały jeden do jednego z S. spontaneum. Jak wspomniano wcześniej w naszym badaniu mapowania genetycznego, Sb08 został podzielony na dwa segmenty, które połączyły się z segmentami Sb02 i segmentami Sb09 w S. spontaneum; Sb05 został podzielony na dwa segmenty, które połączyły się z segmentami Sb06 i segmentami Sb07 S. spontaneum. Dlatego sondy odpowiadające Sb08 i Sb05 nie były specyficzne dla pojedynczych chromosomów S. spontaneum, podczas gdy inne sondy odpowiadające pozostałym ośmiu chromosomom sorgo mogą być stosowane jako sondy cytogenetyczne BAC-FISH specyficzne dla chromosomu dla S. spontaneum. Zatem badane sondy BAC oparte na ośmiu chromosomach sorgo (tabela 2) były wystarczające do identyfikacji chromosomów przy użyciu BAC-FISH. Chromosom satelitarny znaleziono na jednym chromosomie S. spontaneum (chromosom 3) (Ha i wsp., 1999). Wcześniej jednoczesne badanie FISH ujawniło, że sygnały 45S rDNA były zlokalizowane na wtórnych zwężeniach chromosomów satelitarnych w chromosomie 3 z klonu S. spontaneum pochodzącego z hodowli pylników (AP85-361) (Ha i wsp., 1999). W tym badaniu BAC-2, które odpowiadają rDNA Sb03 i 45s, zostały zmapowane do tej samej lokalizacji chromosomu S. spontaneum, co dodatkowo potwierdza wcześniejsze ustalenia (Ha i wsp., 1999). Nasze wyniki dostarczyły również bezpośrednich dowodów na to, że chromosom 3 S. spontaneum nazwany w poprzednim badaniu jest homologiczny do Sb03.
Wniosek
W tym badaniu opracowaliśmy specyficzne chromosomy BAC z S . spontaneum jako krok w kierunku opracowania prostej i powtarzalnej metody identyfikacji chromosomów z wykorzystaniem markerów cytogenetycznych BAC-FISH, potwierdzającej możliwość wyizolowania specyficznych chromosomów BAC na podstawie genomu sorgo w celu skonstruowania fizycznej mapy trzciny cukrowej. Przedstawiamy również pierwszy cytogenetyczny dowód na rearanżację między chromosomami między sorgo i S. spontaneum. Stworzenie systemu technologii BAC-FISH dla trzciny cukrowej otwiera nowe możliwości i możliwości dla badań molekularnej cytogenetyki trzciny cukrowej, w tym analizy kariotypu, lokalizacji genów i budowy mapy fizycznej. Te wyniki są niezbędne do złożenia genomu S. spontaneum wymaganego do sekwencjonowania całego genomu.
Wkład autorów
GD, JS i JZ stworzyli badanie i zaprojektowali eksperyment. GD, JS, QZ, JW, QY, RM, KW i JZ przeprowadzili eksperymenty i przeanalizowali dane. GD i JZ napisali rękopis. Wszyscy autorzy przeczytali i zatwierdzili ostateczny artykuł.
Finansowanie
Ten projekt był wspierany przez granty z programu 863 (2013AA102604), NSFC (31201260), Program for New Century Excellent Talents in Fujian Province University oraz fundusze z Fujian Agriculture and Forestry University.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek powiązań handlowych lub finansowych można to zinterpretować jako potencjalny konflikt interesów.
Podziękowania
Dziękujemy dr Xintan Zhang za zebranie sekwencji BAC i dr Irene Lavagi za redagowanie wersji angielskiej.
Materiały dodatkowe
Dhont, A. (2005). Odkrywanie struktury genomu poliploidów przy użyciu FISH i GISH; przykłady trzciny cukrowej i banana. Cytogenet. Genome Res. 109, 27–33. doi: 10.1159 / 000082378
PubMed Abstract | Pełny tekst CrossRef | Google Scholar
Piperidis, G., D’Hont, A. i Hogarth, D. M. (2001). „Analiza składu chromosomów różnych międzygatunkowych hybryd Saccharum przez genomową in situ (hybrydyzację) (GISH)”, w: Proceedings of the International Society of Sugar Cane Technologists, XXIV Congress (Brisbane, QLD: ASSCT), 565–566.
Google Scholar
